Projekte Systembiologie Mikrobieller Gemeinschaften

Untersuchungen von Genotyp zu Phänotyp bei Bakterien

Da die Sequenzierung des gesamten Genoms von Krankheitserregern bald zur Routine in den Kliniken gehören wird, wird die für die Forschung verfügbare Datenmenge zwangsläufig Stichprobengrößen erreichen, die statistische Genetikansätze zulassen. Dies wiederum bedeutet, dass wir uns allein auf die Genomsequenzen der Erreger stützen können, um die genetischen Determinanten klinisch relevanter Phänotypen wie Pathogenität, Virulenz und Antibiotikaresistenz zu identifizieren, die dann mit vorwärtsgenetischen Assays validiert werden können. Wir erreichen dieses Ziel, indem wir statistische Genetikmethoden nutzen, die wir zuvor entwickelt haben, und wir wenden sie auf Arten wie Escherichia coli und Pseudomonas aeruginosa an, mit bereits sehr nützlichen Ergebnissen.
Referenzen:
- Galardini M, Koumoutsi A, Herrera-Dominguez L, Cordero Varela JA, Telzerow A, Wagih O, Wartel M, Clermont O, Denamur E, Typas A, Beltrao P (2017) Phenotype inference in an Escherichia coli strain panel. Elife 6, p.e31035.
Lees, J. A., Galardini, M., Bentley, S. D., Weiser, J. N., & Corander, J. (2018). Pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics, 34(24), 4310-4312.
Galardini, M., Clermont, O., Baron, A., Busby, B., Dion, S., Schubert, S., ... & Denamur, E. (2020). Major role of iron uptake systems in the intrinsic extra-intestinal virulence of the genus Escherichia revealed by a genome-wide association study. PLoS genetics, 16(10), e1009065. - Denamur, E., Condamine, B., Esposito-Farèse, M., Royer, G., Clermont, O., Laouenan, C., … & Galardini, M.* (2022). Genome wide association study of human bacteremia Escherichia coli isolates identifies genetic determinants for the portal of entry but not fatal outcome. PLoS Genet. 2022;18(3):e1010112.
- Neubauer, H., & Galardini, M. (2023). Improved interpretability of bacterial genome-wide associations using gene cluster centric k-mers. BioRxiv. 10.1101/2023.04.11.536385
- Burgaya, J.*, Marin, J.*, Royer, G., Condamine, B., Gachet, B., Clermont, O., Jaureguy, F., Burdet, C., Lefort, A., Lastours, V. de, Denamur, E.*, Galardini, M.*, Blanquart, F.* (2023). The bacterial genetic determinants of Escherichia coli capacity to cause bloodstream infections in humans. PLOS Genetics, 19(8), e1010842.
Prüfung der Auswirkungen des genetischen Hintergrunds auf die antimikrobielle Resistenz

Populationsgenetische Studien haben in zunehmendem Maße den Einfluss des genetischen Hintergrunds auf die Auswirkungen von Mutationen auf die Fitness gezeigt, was bedeutet, dass unterschiedliche Anpassungspfade zwischen Stämmen, die zur selben Art gehören, zugänglich sein könnten. Diese große genetische Variabilität zwischen den Stämmen wirkt sich wahrscheinlich auf die Fähigkeit aus, mehr oder weniger leicht eine antimikrobielle Resistenz (AMR) zu entwickeln. Darüber hinaus ist bekannt, dass die Interaktion zwischen genetischen Varianten (d. h. epistatische Effekte) die Anpassung beeinflusst und zu einer "zerklüfteten" Fitnesslandschaft führt, die für jeden genetischen Hintergrund sehr spezifisch ist. Infolgedessen können bestimmte Anpassungspfade für einen Stamm nicht zugänglich sein, während andere begünstigt werden. Wir setzen die automatisierte Laborevolution (ALE) bei natürlichen E. coli-Isolaten ein, um die Wechselwirkung zwischen dem genetischen Hintergrund und der Evolution von AMR zu verstehen.
Referenzen:
-
Galardini M, Busby BP, Vieitez C, Dunham AS, Typas A, Beltrao P (2019) The impact of the genetic background on gene deletion phenotypes in Saccharomyces cerevisiae. Mol Syst Biol 15(12): e8831.
Pangenomweite Vorhersage der Genfunktion
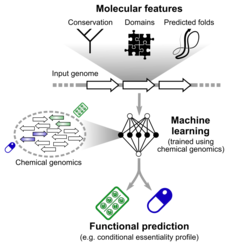
Mit dem Aufkommen der Hochdurchsatz-Sequenzierung ist es möglich geworden, die Genomsequenz von Hunderten von Bakterienisolaten mit begrenzten Kosten zu erhalten. Wir wissen heute, dass sich bei Arten wie E. coli einzelne Stämme in ihrem Gengehalt um bis zu 60 % unterscheiden. Es ist bekannt, dass die Gene mit geringem Erhaltungsgrad, auch akzessorische Gene genannt, zum Überleben in spezialisierten Nischen beitragen; für viele von ihnen ist jedoch nicht einmal eine umfassende funktionelle Charakterisierung verfügbar, wobei die Aussichten für Mitglieder des menschlichen Mikrobioms noch schlechter sind. Chemische Genomikansätze können verwendet werden, um die Funktionen dieser Gene zu rekonstruieren, sind aber aus Kosten- und Arbeitsgründen auf einige Dutzend Arten beschränkt. Wir verwenden computergestützte Ansätze wie maschinelles Lernen, das auf der Fülle der für Modellorganismen verfügbaren Daten trainiert wurde, und nutzen aus Nukleotidsequenzen extrahierte Merkmale, um die derzeitigen Methoden zur Funktionsvorhersage zu verbessern.