Projects Systems Biology of Microbial Communities

Genotype to phenotype studies in bacteria

As pathogen whole genome sequencing is poised to become standard routine in the clinics, the amount of data available for research is bound to reach sample sizes that make it amenable to statistical genetics approaches. This in turn means that we can rely solely on the genome sequences of the pathogens to identify the genetic determinants of clinically relevant phenotypes such as pathogenicity, virulence and antimicrobial resistance, which can then be validated with forward genetic assays. We are achieving this objective by leveraging statistical genetics methods we have previously developed and we are applying them to species such as Escherichia coli and Pseudomonas aeruginosa, already with very useful results.
References:
- Galardini M, Koumoutsi A, Herrera-Dominguez L, Cordero Varela JA, Telzerow A, Wagih O, Wartel M, Clermont O, Denamur E, Typas A, Beltrao P (2017) Phenotype inference in an Escherichia coli strain panel. Elife 6, p.e31035.
Lees, J. A., Galardini, M., Bentley, S. D., Weiser, J. N., & Corander, J. (2018). Pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics, 34(24), 4310-4312. - Galardini, M., Clermont, O., Baron, A., Busby, B., Dion, S., Schubert, S., ... & Denamur, E. (2020). Major role of iron uptake systems in the intrinsic extra-intestinal virulence of the genus Escherichia revealed by a genome-wide association study. PLoS genetics, 16(10), e1009065.
- Denamur, E., Condamine, B., Esposito-Farèse, M., Royer, G., Clermont, O., Laouenan, C., … & Galardini, M.* (2022). Genome wide association study of human bacteremia Escherichia coli isolates identifies genetic determinants for the portal of entry but not fatal outcome. PLoS Genet. 2022;18(3):e1010112.
- Neubauer, H., & Galardini, M. (2023). Improved interpretability of bacterial genome-wide associations using gene cluster centric k-mers. BioRxiv. 10.1101/2023.04.11.536385
- Burgaya, J.*, Marin, J.*, Royer, G., Condamine, B., Gachet, B., Clermont, O., Jaureguy, F., Burdet, C., Lefort, A., Lastours, V. de, Denamur, E.*, Galardini, M.*, Blanquart, F.* (2023). The bacterial genetic determinants of Escherichia coli capacity to cause bloodstream infections in humans. PLOS Genetics, 19(8), e1010842.
Testing the impact of genetic backgrounds on antimicrobial resistance

Population genetics studies have increasingly shown the influence of genetic background on mutations’ fitness effects, which implies that different adaptation trajectories might be accessible across strains belonging to the same species. This large genetic variability across strains is likely to affect the ability to more or less readily develop antimicrobial resistance (AMR). In addition, the interaction between genetic variants (i.e. epistatic effects) are known to influence adaptation, leading to a “rugged” fitness landscape that is highly specific to each genetic background. As a result, certain adaptation trajectories might not be accessible to a strain, while others may be favoured. We are using Automated Laboratory Evolution (ALE) across E. coli natural isolates to understand the interaction between genetic backgrounds and evolution of AMR.
References:
- Galardini M, Busby BP, Vieitez C, Dunham AS, Typas A, Beltrao P (2019) The impact of the genetic background on gene deletion phenotypes in Saccharomyces cerevisiae. Mol Syst Biol 15(12): e8831.
Pangenome wide prediction of gene function
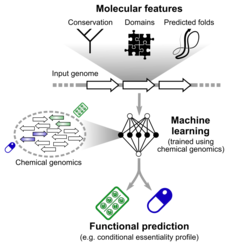
The advent of high-throughput sequencing has resulted in the possibility to obtain the genome sequence of hundreds of bacterial isolates with limited cost. We now know that for species such as E. coli individual strains differ for up to 60% in their gene content. Those genes with low conservation, also called accessory genes, are known to contribute to survival in specialized niches; even a broad functional characterization is however not available for many of them, with an even worse outlook for members of the human microbiome. Chemical genomics approaches can be used to reconstruct the functions of these genes, but are limited to a few tens of species because of cost and labour constraints. We are using computational approaches such as machine learning trained on the wealth of data available for model organisms and using features extracted from nucleotide sequences to improve the current function prediction methods.